Tesi doctoral de Frederico de Lacerda Ferreira Dos Santos: "Density Functional Theory to the Rescue of Transition-metal Chemistry". Direcció: Dr. Marcel Swart. Departament de Química
 Il·lustració autor tesi
Il·lustració autor tesi
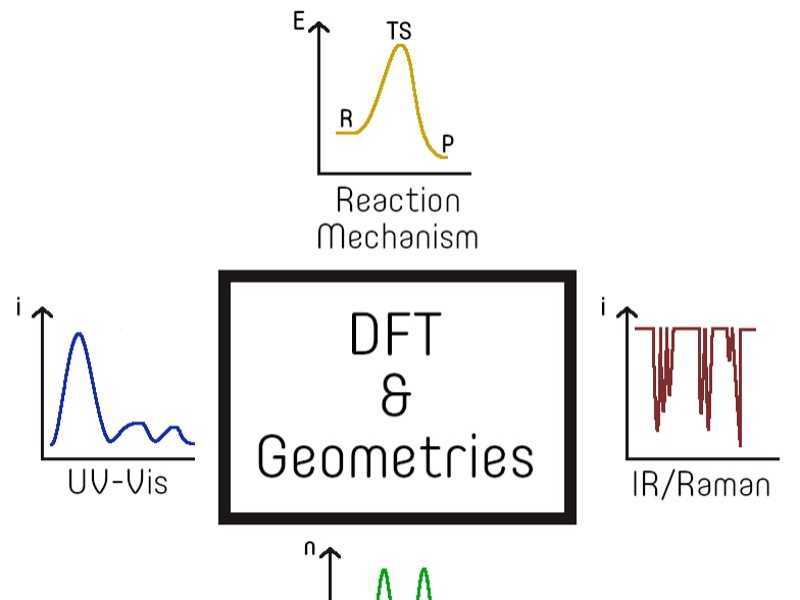
La teoria del funcional de densitat (DFT, per les seves sigles en anglès) va sorgir fa aproximadament 60 anys, dotant els químics d'un marc computacional poderós per simular el comportament dels sistemes químics en un entorn virtual. Malgrat la seva àmplia utilitat, els mètodes de DFT han trobat dificultats per modelar adequadament la reactivitat dels complexos de metalls de transició, a causa de, p. ex., les seves estructures electròniques de capa oberta, caràcter multi referència i les conseqüències associades. No obstant això, aquests complexos juguen un paper crucial com a components essencials de materials amb funcionalitat excepcional, permetent dur a terme reaccions complexes que, d'altra manera, serien extremadament desafiants, similars a les facilitades pels cofactors enzimàtics.
D'una manera sorprenent, aquest treball demostra de manera fortuïta la capacitat dels mètodes DFT per superar els obstacles existents en la química dels metalls de transició. Al Capítol 4, explorem la relació entre les freqüències vibracionals, l'estructura i les propietats magnètiques dels complexos de di ferro amb ponts oxo, semblants al cofactor present en l'enzim monooxigenasa soluble en metà. Al Capítol 5, utilitzem tècniques basades en DFT per localitzar els electrons en sistemes π altament deslocalitzats de metal·lo porfirines, aclarint la seva influència en la banda Soret d'aquests complexos. El Capítol 6 posa en relleu la importància de l'estimació inicial en els estudis de reactivitat, ja que ens enfrontem a desafiaments per obtenir la reactivitat desitjada de l'activació de l'enllaç C-H en un complex níquel-halur, probablement a causa d'un mínim espuri en la superfície d'energia potencial obtinguda. Al Capítol 7, demostrem la utilitat dels càlculs de DFT dependents del temps per obtenir amb precisió els espectres UV-Visibles de complexos de ferro-oxo de València alta, permetent la seva identificació. Finalment, el Capítol 8 explora l'activació cooperativa del nitrogen molecular amb un complex de reni de metall de transició i àcids de Lewis, explicant els fenòmens observats des del punt de vista de la teoria dels orbitals moleculars.
En general, aquesta tesi pretén posar de manifest la vitalitat i la rellevància contínua dels mètodes de DFT en la recerca actual. Emprant aquests mètodes per esbrinar problemes químics, som conscients de les seves limitacions i sovint recorrem a solucions alternatives per superar aquests desafiaments.
Lectura de la tesi: 09/10/23, Sala de Graus de la Facultat de Lletres (informació extreta de l’Agenda activitats de la web Escola de Doctorat)
Notícies relacionades